Researchers describe molecular mechanism involved in Huntington’s neurodegeneration
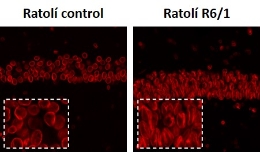
Protein alteration in the family of lamins causes several diseases, known as laminopathies, such as progeria or precocious ageing. A study in which UB researchers have taken part states that alterations in the levels of one of these proteins, lamin B1, contribute to the degeneration of different brain neuronal populations in Huntington’s disease. Caused by a mutation in the huntingtin gen, this pathology features involuntary movements, cognitive deficit and psychiatric disorders, and has no cure yet. According to the study, published in the journal EMBO Molecular Medicine, these results open new therapeutic pathways for the treatment of this disease, since research shows pharmacological normalization of levels of lamin B1 improve the cognitive symptoms in a transgenic model of the disease.
The study counts on the participation of researchers of the Institute of Neurosciences of the University of Barcelona (UBNeuro), the Biomedical Research Networking Center in Neurodegenerative Diseases (CIBERNED), and the August Pi i Sunyer Biomedical Research Institute (IDIBAPS), and is led by Esther Pérez-Navarro, tenured university lecturer at the Faculty of Medicine and Health Sciences.
Among the participants in the study are also researchers from the Center for Genomic Regulation (CRG), Pompeu Fabra University, Vall d’Hebron Institute of Oncology and the University of Cambridge (United Kingdom).
An innovative technique to analyze cell nucleus
The level increase of lamin B1, a protein in the cell nucleus, causes a rare disease, the autosomal dominant leukodystrophy, which features motor and cognitive deficits and demyelination in the central nervous system. In this study, researchers analyzed the impact at a physiologic, transcriptomic and epigenetic level of these alterations in Huntington’s disease. To do so, they used the experiments in a transgenic mice model of the pathology and post-mortem brain samples from patients. One of the most relevant aspects of the study is the development of an innovative technique, the fluorescence-activated nuclear suspension imaging (FANSI) to identify and analyze the neuron nucleus of specific populations in the brain. “This technique could be used by other research groups”, notes Esther Pérez-Navarro.
“Moreover —the researcher adds—, we have applied the ChIP-sequencing technique in collaboration with other research groups experts in this technique and the interpretation of results: the groups led by Mashami Narita, from the University of Cambridge, and Luciano Di Croce, from CRG. This allowed us to analyze how the alteration of levels in lamin B1 can involve changes in gene transcription”.
Later, researchers gave the mice betulinic acid, a drug that can partially restore the levels of lamin B1, to test the effect in the mice models of the disease. Normalization of levels of this protein in the nucleus of neuronal populations palliated motor and cognitive disfunctions in animals. According to the researchers, these results show that the level increase of lamin B1 contributes to the alteration of the nuclear function of specific neurons in the brain in Huntington’s disease. Pérez-Navarro states that “to date, we did not know about the alteration of this protein being involved in Huntington’s physiopathology”, and she questions whether this alteration could be involved in the neuronal degeneration of other diseases, such as Alzheimer’s and Parkinson’s.
First step to design new drugs
The identification of this molecular mechanism sheds light on the design of new drugs to treat a disease with no cure yet. “Betulinic acid used in this research has other effects and therefore we need to identify specific drugs that target the modulation of lamin B1 levels”, notes the researcher.
Moreover, this study could have implications in the identification of new biomarkers of the disease. In this sense, the future goal of the researchers is to determine whether lamin B levels are also altered in cells outside the brain, such as blood cells and fibroblasts (skin cells). “The benefits of this study in a disease such as Huntington’s, associated with a genetic mutation, is that we can analyze these chanhes in carriers that have no symptoms yet and monitor it over time. To carry out these studies, we are working with the Department of Movement Disorders (Jesús Pérez and Jaime Kulisevsky) in Hospital de la Santa Creu i Sant Pau, a distinguished center regarding this disease”, concludes the researcher.
Reference article:
Alcalá-Vida, R.; Garcia-Forn, M.; Castany-Pladevall, C.; Creus-Muncunill, J.; Ito, Y.; Blanco, E.; Golbano. A.; Crespí-Vázquez, K.; Parry, A.; Slater, G.; Samarajiwa, S.; Peiró, S.; Di Croce, L.; Narita, M. i Pérez-Navarro, E. “Neuron type-specific increase in lamin B1 contributes to nuclear dysfunction in Huntington’s disease”. EMBO Mol Med. December, 2020. Doi: https://doi.org/10.15252/emmm.202012105.
No Comments