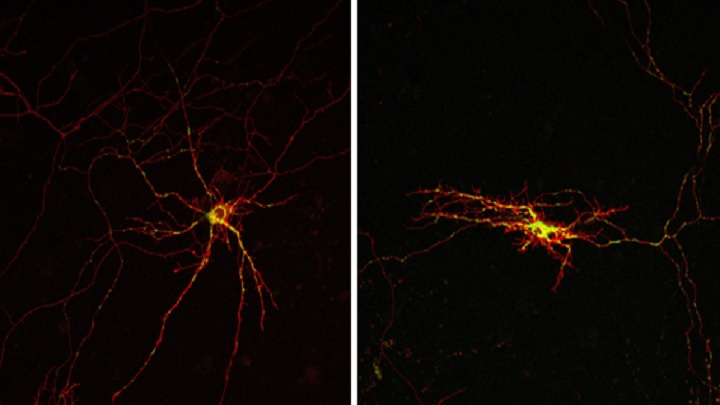
This study finds a molecular mechanism related to neuronal death
The human brain is an organ that takes nearly from 20 to 25% of the energy the body needs. This high energy demand for neuronal functions depends on the transport and precise distribution of mitochondria — the energy-generating cell organelles — in each neuron. Now, a study published in the journal Science Signaling has identified, for the first time, a molecular complex that regulates the transport of mitochondria within neurons and neuronal death. The discovery of the complex, exclusively present in the most evolved mammals, could help to locate new therapeutic targets against neurodegenerative diseases such as Parkinson’s, neuromuscular diseases or even some types of tumours.
The study, conducted on animal models and cell cultures, is led by Professor Eduardo Soriano, from the University of Barcelona and the Institute of Neurosciences of the UB (UBneuro), and the Biomedical Research Networking Centre on Neurodegenerative Diseases (CIBERNED), and researcher Anna María Aragay, member of the Spanish National Research Council (CSIC) and the Institute of Molecular Biology of Barcelona (IBMB-CSIC).
The article, whose first authors are Ismael Izquierdo-Villalba (IBMB-CSIC), Serena Mirra and Yasmina Manso (UB-CIBERNED), includes the participation of Adolfo López de Munain, from the University Hospital of Donostia, Xavier Navarro, from the Autonomous University of Barcelona (UAB), both members of CIBERNED, and José Antonio Enríquez, collaborator at the Biomedical Networking Research on Fragility and Healthy Ageing (CIBERFES) and the National Centre of Cardiovascular Research Carlos III (CNIC).
Bringing energy for neuronal functions
“In neurons, the transport process of mitochondria is determining, since these organelles must be present along all axons and dendrites — neuron extensions — to provide energy to the neurotransmission and the neuronal functions, processes that require a lot of energy. This great consumption depends on a specific and precise distribution of mitochondria within neurons”, notes Soriano, co-director of the study and member of the Department of Cell Biology, Physiology and Immunology at the UB’s Faculty of Biology.
The study reveals the Alex3/Gαq mitochondrial complex interacts with the mitochondria machinery to distribute and transport these cell organelles along the neurons’ axons and dendrite. This process depends on the interaction of the Gq protein with the Alex3 mitochondrial protein.
“For the first time, we found that the Alex3/Gαq is essential not only for the transport and mitochondrial function, but also for neuronal physiology, movement control and neuronal viability. If this system is inactivated — for instance, in mice with a specific deficiency of the Alex3 protein in the central nervous system — the mitochondrial trafficking is reduced, there is less dendritic and axonal arborizations and this causes motor deficits and even neuronal death”, says Aragay, co-director of the study.
The authors of the study had previously described in other articles that the Alex3 and Gαq proteins regulated mitochondrial transport. However, they did not know how these interacted or what molecular mechanisms took part in the process.
The interaction of the Alex3/Gαq mitochondrial complex is regulated through the G protein-coupled receptors (GPCR), according to the study. These receptors have many molecules — neurotransmitters, hormones, cannabinoids, etc. — with different functions in the organism.
“The activation of GPCRs not only alters the mitochondrial distribution but also its function, and as a notable effect, the neuronal growth and viability. Our study suggests that, in general, these molecules that interact with these receptors could regulate several aspects of the mitochondrial biology through the GPCR”, note the experts.
Controlling receptors to fight human diseases
Although the action mechanisms are not well known yet, it seems that different functions played by the Alex3 protein could be associated with many pathologies. For instance, it appears that deletions — loss of a DNA fragment — of the Alex3 facilitate the development of certain tumours (epithelial cancers). In other cases, the deletion or inhibition of its expression has a protective effect on certain tumours (liver cancers).
Apart from its association with cancer, some genic variants of the Alex3 protein and its genic family are also related to neurodegenerative diseases — especially Parkinson’s —, sleep apnoea and metabolic diseases.
“The fact that inactivating mutations have not been identified in the databanks of thousands of human genomes would indicate that the Alex3 gene has a relevant function. Its total loss is not viable in the organism, and it would be found as a somatic mutation in tumours”, says Professor Gemma Marfany, co-author of the study and member of the UB’s Department of Genetics, Microbiology and Statistics, the Institute of Biomedicine of the UB (IBUB) and the Rare Diseases Networking Biomedical Research Centre (CIBERER).
“Moreover, mutations in the gene that codes for Gαq in humans lead to motor disorders, cognitive deficits, intellectual disability and epilepsy”, notes Aragay. The authors highlight that these data show the relevance of the identified complex for neuronal function.
“Being able to control mitochondrial biology from outside the cell via GPCR receptors is a great advantage. Currently, many specific molecules activate or inhibit these receptors, so it is important to explore the possibility of controlling the localization and biology of mitochondria in diseases where there is a deficit of these organelles (e.g. mitochondrial or neuromuscular diseases), or in pathologies where inhibition of metabolism has positive therapeutic effects (e.g. cancer)”, the team concludes.
Reference article:
Izquierdo-Villalba, I.; Mirra, S. et al. “A mammalian-specific Alex3/Gαq protein complex regulates mitochondrial trafficking, dendritic complexity, and neuronal survival”. Science Signaling, February 2024. DOI: 10.1126/scisignal.abq1007
Sorry, the comment form is closed at this time.